Cálculo de Carga con Espresso
Como en el caso de las bandas, acá partimos de un exemplo pero de PP (en vez de PW).

Primero describiré brevemente la corrida debido a que el script es un poco largo. Son 4 pasos.
- Corrida SCF de Si. Se hace una corrida normal para el silicio. En este caso, PREFIX=si.
- Se hace el post-processing para la densidad de carga.
- &inputpp: hay dos variables, filplot y plot_num que vale la pena comentar. Primero, plot_num= 0 significa que se salvara la carga en filplot. Claro que filplot es el archivo donde se guardara. La variable plot_num puede por supuesto tomar otros valores:
0 = charge 1 = total potential V_bare+V_H + V_xc 2 = local ionic potential 3 = local density of states at e_fermi 4 = local density of electronic entropy 5 = STM images 6 = spin polarization (rho(up)-rho(down)) 7 = |psi|^2 8 = electron localization function (ELF) 9 = planar average of all |psi|^2 10 = integrated local density of states (ILDOS) from emin to emax (emin, emax in eV) if emax is not specified, emax=E_fermi 11 = the V_bare + V_H potential 12 = the electric field potential 13 = the noncollinear magnetization. - &plot. Aquí hay que mencionar varias. nfile=1 es default e indica que se genera 1 archivo. Luego, filepp(1) es filplot a menos que nfile fuera distinto de 1. Aquí sólo interesa este caso. weight(1)=1 tambien es dafault. La siguiente variable iflag =2 dice que es una gráfica 2D. Otros valores para iflag son:
0 1D plot of the spherical average 1 1D plot 2 2D plot 3 3D plot 4 2D polar plot on a sphere
Relacionada con la anterior, esta la variable output_format=2 que hace un formato adecuado para el programa plotrho.x. Otros valores de esta variable son
(ignored on 1D plot) 0 format suitable for gnuplot (1D) 1 format suitable for contour.x (2D) 2 format suitable for plotrho (2D) 3 format suitable for XCRYSDEN (1D, 2D, 3D) 4 format suitable for gOpenMol (3D) (formatted: convert to unformatted *.plt) 5 format suitable for XCRYSDEN (3D) 6 format as gaussian cube file (3D) (can be read by many programs)
La siguiente variable es fileout=’si.rho.dat’ que indica el nombre del archivo donde se guardará la gráfica. Dependiendo del valor de iflag, se definen lineas, planos o volumenes. En este caso, se definen los vectores ortogonales e1=(1,1,0) y e2=(0,0,1) que representa el plano [110]. Junto con estos vectores, también se puede definir el origen y el numero de punto en el mallado.
- Despues de estos pasos, se vuelve a grabar la carga pero en formatos para gnuplot (aparentemente con output_format = 7 ) y también se calculan bandas.
- &inputpp: hay dos variables, filplot y plot_num que vale la pena comentar. Primero, plot_num= 0 significa que se salvara la carga en filplot. Claro que filplot es el archivo donde se guardara. La variable plot_num puede por supuesto tomar otros valores:
A continuación se muestra el script para correr el ejemplo en el cluster:
———————————————————————–
#!/bin/sh
#PBS -l nodes=1:ppn=10 -q supermicro
#PBS -N batio3
#PBS -e .ERROR
#PBS -o .OUT
CODE=/usr/local/cluster_programs/espresso/
# run from directory where this script is
#cd `echo $0 | sed ‘s/\(.*\)\/.*/\1/’` # extract pathname
cd «$PBS_O_WORKDIR»
PW_COMMAND=pw.x
# Run the Application
export OMP_NUM_THREADS=$PBS_NP
PSEUDO_DIR=`.`
TMP_DIR=`.`
# required executables and pseudopotentials
BIN_LIST=»pw.x pp.x plotrho.x bands.x plotband.x»
PSEUDO_LIST=»Si.pz-vbc.UPF»
# check for gnuplot
GP_COMMAND=`which gnuplot 2>/dev/null`
# check for directories
PP_COMMAND=pp.x
PLOTRHO_COMMAND=plotrho.x
BANDS_COMMAND=bands.x
PLOTBAND_COMMAND=plotband.x
# self-consistent calculation
cat > si.scf.in << EOF
&control
calculation=’scf’
restart_mode=’from_scratch’,
prefix=’si’
pseudo_dir = ‘.’,
outdir=’.’
/
&system
ibrav= 2, celldm(1)= 10.2, nat= 2, ntyp= 1,
ecutwfc =18.0
/
&electrons
conv_thr = 1.0d-8
mixing_beta = 0.7
/
ATOMIC_SPECIES
Si 28.086 Si.pz-vbc.UPF
ATOMIC_POSITIONS
Si 0.00 0.00 0.00
Si 0.25 0.25 0.25
K_POINTS
10
0.1250000 0.1250000 0.1250000 1.00
0.1250000 0.1250000 0.3750000 3.00
0.1250000 0.1250000 0.6250000 3.00
0.1250000 0.1250000 0.8750000 3.00
0.1250000 0.3750000 0.3750000 3.00
0.1250000 0.3750000 0.6250000 6.00
0.1250000 0.3750000 0.8750000 6.00
0.1250000 0.6250000 0.6250000 3.00
0.3750000 0.3750000 0.3750000 1.00
0.3750000 0.3750000 0.6250000 3.00
EOF
$PW_COMMAND < si.scf.in > si.scf.out
# post-processing for charge density
cat > si.pp_rho.in << EOF
&inputpp
prefix = ‘si’
outdir = ‘.’
filplot = ‘sicharge’
plot_num= 0
/
&plot
nfile = 1
filepp(1) = ‘sicharge’
weight(1) = 1.0
iflag = 2
output_format = 2
fileout = ‘si.rho.dat’
e1(1) =1.0, e1(2)=1.0, e1(3) = 0.0,
e2(1) =0.0, e2(2)=0.0, e2(3) = 1.0,
nx=56, ny=40
/
EOF
$PP_COMMAND < si.pp_rho.in > si.pp_rho.out
# plotrho
cat > si.plotrho.in << EOF
si.rho.dat
si.rho.ps
n
0 0.09 6
EOF
$PLOTRHO_COMMAND < si.plotrho.in > si.plotrho.out
# post-processing for charge density
cat > si.pp_rho_new.in << EOF
&inputpp
prefix = ‘si’
outdir = ‘.’
filplot = ‘sicharge’
plot_num= 0
/
&plot
nfile = 1
filepp(1) = ‘sicharge’
weight(1) = 1.0
iflag = 2
output_format = 7
fileout = ‘si.rho_new.dat’
e1(1) =1.0, e1(2)=1.0, e1(3) = 0.0,
e2(1) =0.0, e2(2)=0.0, e2(3) = 1.0,
nx=141, ny=100
/
EOF
$ECHO
$ECHO » running pp.x to do another 2-d plot of the charge density…\c»
$PP_COMMAND < si.pp_rho_new.in > si.pp_rho_new.out
cat > gnuplot.tmp <<EOF
#!$GP_COMMAND
#
set term png font «,18» enh size 1000,707
set pm3d
set palette model HSV functions gray*0.75, 1, 0.9
set view 0,0
#
alat=10.2
set xra[0:1.4142136*alat]
set yra [0.:alat]
set xtics out nomirror
set ytics axis in offset -4.0,0 nomirror
set label «r (a.u)» at 6.8,-2.2 center
set label «r (a.u)» at -1.7,5.0 rotate by 90 center
unset ztics
unset key
set colorbox
#
set out ‘si.charge.png’
set title «Si charge»
splot ‘si.rho_new.dat’ u 1:2:3 w pm3d
EOF
$GP_COMMAND < gnuplot.tmp
#rm gnuplot.tmp
cat > gnuplot1.tmp <<EOF
#!$GP_COMMAND
set view map
set size square
unset surface
unset clabel
set contour
set dgrid3d 100,141
set cntrparam cubicspline
set table
#
# Define here the countour values. Each set of countours will have the same
# color and is written in a different file
#
set cntrparam levels discrete 0.01,0.02,0.03
set output «table.dat»
splot ‘si.rho_new.dat’ using 1:2:3 w l
set cntrparam levels discrete 0.04,0.05,0.06
set output «table1.dat»
splot ‘si.rho_new.dat’ using 1:2:3 w l
set cntrparam levels discrete 0.07,0.08
set output «table2.dat»
splot ‘si.rho_new.dat’ using 1:2:3 w l
#
unset table
#
# Now define a postcript terminal
#
set encoding iso_8859_15
set terminal postscript enhanced solid color «Helvetica» 20
set output «si.contour.ps»
#
# prepare the countour graph
#
set size ratio 1./1.4142
set key off
alat=10.2
set border lw 3
set label «Si» at 10.6,8.7
set xrange [0:1.4142136*alat]
set yrange [0:alat]
set xlabel «r (a.u.)»
set ylabel «r (a.u.)»
#
# Set contour labels
#
dato=»0.08″
set obj 9 rect at 6.,1.3 size char strlen(dato)*0.6, char 0.6
set obj 9 fillstyle solid noborder front
set label at 6.,1.3 dato front center font «Helvetica,12» tc rgb «blue»
dato=»0.07″
set obj 10 rect at 7.9,1.3 size char strlen(dato)*0.6, char 0.6
set obj 10 fillstyle solid noborder front
set label at 7.9,1.3 dato front center font «Helvetica,12» tc rgb «blue»
dato=»0.06″
set obj 11 rect at 3.3,1.0 size char strlen(dato)*0.6, char 0.6
set obj 11 fillstyle solid noborder front
set label at 3.3,1.0 dato front center font «Helvetica,12» tc rgb «green»
dato=»0.01″
set obj 12 rect at 3.6,6.0 size char strlen(dato)*0.6, char 0.6
set obj 12 fillstyle solid noborder front
set label at 3.6,6.0 dato front center font «Helvetica,12» tc rgb «red»
dato=»0.02″
set obj 13 rect at 3.6,5.4 size char strlen(dato)*0.6, char 0.6
set obj 13 fillstyle solid noborder front
set label at 3.6,5.4 dato front center font «Helvetica,12» tc rgb «red»
dato=»0.03″
set obj 14 rect at 3.6,4.9 size char strlen(dato)*0.6, char 0.6
set obj 14 fillstyle solid noborder front
set label at 3.6,4.9 dato front center font «Helvetica,12» tc rgb «red»
dato=»0.04″
set obj 15 rect at 2.3,3.7 size char strlen(dato)*0.6, char 0.6
set obj 15 fillstyle solid noborder front
set label at 2.3,3.7 dato front center font «Helvetica,12» tc rgb «green»
dato=»0.05″
set obj 16 rect at 3.6,3.7 size char strlen(dato)*0.6, char 0.6
set obj 16 fillstyle solid noborder front
set label at 3.6,3.7 dato front center font «Helvetica,12» tc rgb «green»
dato=»0.05″
set obj 17 rect at 7.2,1.9 size char strlen(dato)*0.6, char 0.6
set obj 17 fillstyle solid noborder front
set label at 7.2,1.9 dato front center font «Helvetica,12» tc rgb «green»
dato=»0.01″
set obj 18 rect at 10.8,2.3 size char strlen(dato)*0.6, char 0.6
set obj 18 fillstyle solid noborder front
set label at 10.8,2.3 dato front center font «Helvetica,12» tc rgb «red»
#
# Print the countour
#
plot «table.dat» u 1:2 w l lw 3 lc rgb «red», «table1.dat» u 1:2 w l lw 3 lc rgb «green», «table2.dat» u 1:2 w l lw 3 lc rgb «blue»
EOF
$GP_COMMAND < gnuplot1.tmp
rm gnuplot1.tmp table.dat table1.dat table2.dat
# band structure calculation along high-symmetry lines
cat > si.band.in << EOF
&control
calculation=’bands’
pseudo_dir = ‘.’,
outdir=’.’,
prefix=’si’
/
&system
ibrav= 2, celldm(1) =10.20, nat= 2, ntyp= 1,
ecutwfc =18.0, nbnd = 8,
/
&electrons
/
ATOMIC_SPECIES
Si 28.086 Si.pz-vbc.UPF
ATOMIC_POSITIONS
Si 0.00 0.00 0.00
Si 0.25 0.25 0.25
K_POINTS tpiba_b
5
L 20
gG 20
X 0
1.0 1.0 0.0 30
gG 1
EOF
$PW_COMMAND < si.band.in > si.band.out
# post-processing for band structure
cat > si.bands.in << EOF
&bands
prefix = ‘si’
outdir = ‘.’
filband = ‘sibands.dat’
lsym=.true.,
/
EOF
$BANDS_COMMAND < si.bands.in > si.bands.out
# plotband.x
cat > si.plotband.in << EOF
sibands.dat
-6.0 10
sibands.xmgr
sibands.ps
6.255
1.0 6.255
EOF
$PLOTBAND_COMMAND < si.plotband.in > si.plotband.out
cat > gnuplot1.tmp <<EOF
set encoding iso_8859_15
set terminal postscript enhanced solid color «Helvetica» 20
set output «si.bands.ps»
#
set key off
dim1=-12.5
dim2=6
sqrt32=sqrt(3)*0.5
sqrt2=sqrt(2)
set xrange [0:sqrt2+sqrt32+1]
set yrange [dim1:dim2]
set arrow from sqrt32,dim1 to sqrt32,dim2 nohead lw 2 front
set arrow from sqrt32+1,dim1 to sqrt32+1,dim2 nohead lw 2 front
set arrow from 0,0 to sqrt2+sqrt32+1,0 nohead lw 1 front
set ylabel «Energy (eV)»
set label «Si» at 0.3,5 center
unset xtics
set border lw 2
lpos=dim1-0.45
set label «L» at 0.,lpos center
set label «{/Symbol G}» at sqrt32,lpos center
set label «X» at sqrt32+1,lpos center
set label «{/Symbol G}» at sqrt2+sqrt32+1,lpos center
set label «L_1» at 0.3,-10. center tc lt 1
set label «L_3» at 0.3,-2. center tc lt 2
set label «{/Symbol G}_1» at sqrt32+0.02,-11.5 left
set label «{/Symbol G}_{25}'» at sqrt32+0.02,0.5 left
set label «{/Symbol G}_{15}» at sqrt32+0.02,2.0 left
set label «{/Symbol G}_{2}'» at sqrt32+0.02,4.6 left
set label «{/Symbol D}_1» at sqrt32+0.6,-9.5 center tc lt 1
set label «{/Symbol D}_2′» at sqrt32+0.6,-5.5 center tc lt 2
set label «{/Symbol D}_5» at sqrt32+0.6,-1.7 center tc lt 3
set label «{/Symbol S}_1» at sqrt32+1.7,-11 center tc lt 1
set label «{/Symbol S}_4» at sqrt32+1.7,-6.5 center tc lt 3
set label «{/Symbol S}_2» at sqrt32+1.7,-2.2 center tc lt 2
set label «{/Symbol S}_3» at sqrt32+1.7,4. center tc lt 5
hpos=0.6
set label «C_{3v}» at sqrt32*0.5,dim2+hpos center
set label «O_h» at sqrt32,dim2+hpos center
set label «C_{4v}» at sqrt32+0.5,dim2+hpos center
set label «C_{2v}» at sqrt32+1.0+sqrt2/2,dim2+hpos center
set label «O_h» at sqrt32+1.0+sqrt2,dim2+hpos center
vb_max=6.255
plot «sibands.xmgr.1.1» u 1:(\$2-vb_max) w l lw 3 lt 1 ,\
«sibands.xmgr.1.3» u 1:(\$2-vb_max) w l lw 3 lt 2 ,\
«sibands.xmgr.2.1» u 1:(\$2-vb_max) w l lw 3 lt 1 ,\
«sibands.xmgr.2.4» u 1:(\$2-vb_max) w l lw 3 lt 2 ,\
«sibands.xmgr.2.5» u 1:(\$2-vb_max) w l lw 3 lt 3 ,\
«sibands.xmgr.3» u 1:(\$2-vb_max) w l lw 3 lt 1 ,\
«sibands.xmgr.4.1» u 1:(\$2-vb_max) w l lw 3 lt 1 ,\
«sibands.xmgr.4.2» u 1:(\$2-vb_max) w l lw 3 lt 2 ,\
«sibands.xmgr.4.3» u 1:(\$2-vb_max) w l lw 3 lt 5 ,\
«sibands.xmgr.4.4» u 1:(\$2-vb_max) w l lw 3 lt 3
EOF
$GP_COMMAND gnuplot1.tmp
———————————————————————–
K points

En la figura de arriba se hace el ejemplo de un muestreo de la zona de Brillouin y se muestra la construcción de los pesos «weights».
Voy a describir rapidamente la construcción.
En primer lugar, en la figura hay una equivocación al asignar los k. Lo veremos enseguida.
Si los escribimos en notación de vectores unitarios, podemos escribir:

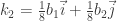
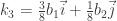
estos representan los puntos en la zona irreducible. Por operaciones de simetría se puede obtener el resto de ellos dando un total de 16 puntos en la Zona de Brillouin. Así, de 
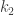

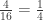
y el peso del

En la suit de quantum espresso, viene un programa para generar los puntos K, que se llama kpoints.x, vamos a usarlo para generar los puntos descritos en el caso de la red cuadrada:
Primero se manda llamar, despiega un mensaje y se le da el número que corresponde a la red estudiada, en nuestro caso sera 1 para cubic p (sc):
$ kpoints.x
***************************************************
* *
* Welcome to the special points world! *
*________________________________________________ *
* 1 = cubic p (sc ) 8 = orthor p (so ) *
* 2 = cubic f (fcc) 9 = orthor base-cent. *
* 3 = cubic i (bcc) 10 = orthor face-cent. *
* 4 = hex & trig p 11 = orthor body-cent. *
* 5 = trigonal r 12 = monoclinic p *
* 6 = tetrag p (st ) 13 = monocl base-cent. *
* 7 = tetrag i (bct) 14 = triclinic p *
***************************************************
bravais lattice >> 1
Enseguida pide el nombre del archivo donde se guarda la información. Por default es mesh_k:
filout [mesh_k] >>nombre_de_archivo
Luego pide el número de puntos k en cada dirección, lo haremos como en el ejemplo de arriba:
mesh: n1 n2 n3 >> 4 4 1
Lo siguente que pide es el shift k1, k2 y k3, si un valor es 0 no hace shift, si es 1 si lo hace. Pondremos 1 en k1 y k2 para centrar en la zona de brillouin. k3 no necesita recorrerse.
mesh: k1 k2 k3 (0 no shift, 1 shifted) >> 1 1 0
Finalmente pregunta si se deben escribir todos los puntos. si se dice «y» se escriben todos, mientras que si es «f» solo se escriben los de la zona irreducible. Haremos los dos casos por separado para comparar.
write all k? [f] >> y
En este caso, regresa el aviso:
# of k-points == 16 of 16
en el caso contrario:
write all k? [f] >>
# of k-points == 3 of 16
Los datos para la zona de brillouin son:
16
1 0.1250000 0.1250000 0.0000000 4.00
2 0.3750000 0.1250000 0.0000000 8.00
3 0.6250000 0.1250000 0.0000000 0.00 2
4 0.8750000 0.1250000 0.0000000 0.00 1
5 0.1250000 0.3750000 0.0000000 0.00 2
6 0.3750000 0.3750000 0.0000000 4.00
7 0.6250000 0.3750000 0.0000000 0.00 6
8 0.8750000 0.3750000 0.0000000 0.00 2
9 0.1250000 0.6250000 0.0000000 0.00 2
10 0.3750000 0.6250000 0.0000000 0.00 6
11 0.6250000 0.6250000 0.0000000 0.00 6
12 0.8750000 0.6250000 0.0000000 0.00 2
13 0.1250000 0.8750000 0.0000000 0.00 1
14 0.3750000 0.8750000 0.0000000 0.00 2
15 0.6250000 0.8750000 0.0000000 0.00 2
16 0.8750000 0.8750000 0.0000000 0.00 1
mientras que para la zona irreducible son:
3
1 0.1250000 0.1250000 0.0000000 4.00
2 0.3750000 0.1250000 0.0000000 8.00
3 0.3750000 0.3750000 0.0000000 4.00
La última columna nos dice cuantos puntos equivalentes hay en la zona completa.
Viendo el PW_forum, encontramos que:
K_POINTS automatic n1 n2 n3 j1 j2 j3 yields a grid of k-points: k(i1,i2,i3) = (i1+j1/2) * g1/n1 + (i2+j2/2) * g2/n2 + (i3+j3/2) * g3/n3 where g1, g2, g3 are the vectors that generate the reciprocal lattice; the indices i1, i2, i3, run from 0 to n1-1, n2-1, n3-1 respectively; j1, j2, j3 can be either 0 (no offset, k=0 in the grid) or 1 (with offset, k=0 not in the grid). What is usually referred to as "Monkhorst-Pack" grid has offset and does not contain k=0 . Of course, only inequivalent k-points are actually used Paolo -- Paolo Giannozzi
En nuestro caso, podemos seguir la receta de esta forma:
n1=4 n2=4 n3= 1, j1=1 j2=1 y j3=0
i1 e i2 = 0,1,2,3 y con i3=0
Los vectores son, entonces:

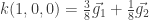

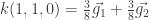
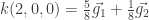
.
.
.

En quantum espresso hay dos formas de hacer un muestreo de la zona de brillouin (ademas de hacer solo el punto gamma).
K_POINTS automatic
nk1 nk2 nk3 sk1 sk2 sk3
genera el mallado en la zona irreducible. Por ejemplo, haciendo
K_POINTS automatic
4 4 4 1 1 1
genera en el output:
number of k points= 10
cart. coord. in units 2pi/alat
k( 1) = ( -0.1250000 0.1250000 0.1250000), wk = 0.0625000
k( 2) = ( -0.3750000 0.3750000 -0.1250000), wk = 0.1875000
k( 3) = ( 0.3750000 -0.3750000 0.6250000), wk = 0.1875000
k( 4) = ( 0.1250000 -0.1250000 0.3750000), wk = 0.1875000
k( 5) = ( -0.1250000 0.6250000 0.1250000), wk = 0.1875000
k( 6) = ( 0.6250000 -0.1250000 0.8750000), wk = 0.3750000
k( 7) = ( 0.3750000 0.1250000 0.6250000), wk = 0.3750000
k( 8) = ( -0.1250000 -0.8750000 0.1250000), wk = 0.1875000
k( 9) = ( -0.3750000 0.3750000 0.3750000), wk = 0.0625000
k( 10) = ( 0.3750000 -0.3750000 1.1250000), wk = 0.1875000
Es equivalente a hacer (el caso default: tpiba)
K_POINTS
10
0.1250000 0.1250000 0.1250000 1.00
0.1250000 0.1250000 0.3750000 3.00
0.1250000 0.1250000 0.6250000 3.00
0.1250000 0.1250000 0.8750000 3.00
0.1250000 0.3750000 0.3750000 3.00
0.1250000 0.3750000 0.6250000 6.00
0.1250000 0.3750000 0.8750000 6.00
0.1250000 0.6250000 0.6250000 3.00
0.3750000 0.3750000 0.3750000 1.00
0.3750000 0.3750000 0.6250000 3.00
Finalmente, si se quiere hacer la estructura de bandas, podemos usar el xcrysden para escoger la trayectoria en la zona de brillouin por los puntos de alta simetría:
Para mostrarlo, podemos usar el archivo de entrada para el cálculo de Si. Con la estructura abierta, vamos a Tools y seleccionamos k-path Selection:


AL hacer click en «ok» abre una ventana para guardar los datos en un archivo. Entre los formatos que da esta el pwscf que guarda la estructura para el input del pw.x. De ahí se puede tomar y pegarse en el archivo:
K_POINTS crystal
50
0.0000000000 0.0000000000 0.0000000000 1.0
0.0416666667 0.0416666667 0.0000000000 1.0
.
.
Cálculo de bandas de Si con Quantum Espresso
Aquí partimos de el ejemplo que viene con el paquete (example01). De las 2 diagonalizaciones sólo se incluye la cg (gradiente conjugado). Se hace un script en el que se generan los archivos de entrada para un cálculo scf y uno de bandas con el programa pw.x
Para hacer más efectiva la corrida y creación de archivos, todo se hace en el archivo job con el siguiente contenido:
————————————-
#!/bin/bash
#PBS -l nodes=1:ppn=10 -q supermicro
#PBS -N batio3
#PBS -e .ERROR
#PBS -o .OUT
CODE=/usr/local/cluster_programs/espresso/
# run from directory where this script is
#cd `echo $0 | sed ‘s/\(.*\)\/.*/\1/’` # extract pathname
cd «$PBS_O_WORKDIR»
PW_COMMAND=pw.x
# Run the Application
export OMP_NUM_THREADS=$PBS_NP
PSEUDO_DIR=`.`
TMP_DIR=`.`
for diago in cg ; do
# self-consistent calculation
cat > si.scf.$diago.in << EOF
&control
calculation = ‘scf’
restart_mode=’from_scratch’,
prefix=’silicon’,
tstress = .true.
tprnfor = .true.
pseudo_dir = ‘.’,
outdir=’.’
/
&system
ibrav= 2, celldm(1) =10.20, nat= 2, ntyp= 1,
ecutwfc =18.0,
/
&electrons
diagonalization=’$diago’
mixing_mode = ‘plain’
mixing_beta = 0.7
conv_thr = 1.0d-8
/
ATOMIC_SPECIES
Si 28.086 Si.pz-vbc.UPF
ATOMIC_POSITIONS
Si 0.00 0.00 0.00
Si 0.25 0.25 0.25
K_POINTS
10
0.1250000 0.1250000 0.1250000 1.00
0.1250000 0.1250000 0.3750000 3.00
0.1250000 0.1250000 0.6250000 3.00
0.1250000 0.1250000 0.8750000 3.00
0.1250000 0.3750000 0.3750000 3.00
0.1250000 0.3750000 0.6250000 6.00
0.1250000 0.3750000 0.8750000 6.00
0.1250000 0.6250000 0.6250000 3.00
0.3750000 0.3750000 0.3750000 1.00
0.3750000 0.3750000 0.6250000 3.00
EOF
# $ECHO » running the scf calculation for Si…\c»
$CODE/$PW_COMMAND < si.scf.$diago.in > si.scf.$diago.out
# $PW_COMMAND < si.scf.$diago.in > si.scf.$diago.out
# check_failure $?
# $ECHO » done»
# band structure calculation along delta, sigma and lambda lines
cat > si.band.$diago.in << EOF
&control
calculation=’bands’
pseudo_dir = ‘.’,
outdir=’.’,
prefix=’silicon’
/
&system
ibrav= 2, celldm(1) =10.20, nat= 2, ntyp= 1,
ecutwfc =18.0, nbnd = 8,
/
&electrons
diagonalization=’$diago’
/
ATOMIC_SPECIES
Si 28.086 Si.pz-vbc.UPF
ATOMIC_POSITIONS
Si 0.00 0.00 0.00
Si 0.25 0.25 0.25
K_POINTS
28
0.0 0.0 0.0 1.0
0.0 0.0 0.1 1.0
0.0 0.0 0.2 1.0
0.0 0.0 0.3 1.0
0.0 0.0 0.4 1.0
0.0 0.0 0.5 1.0
0.0 0.0 0.6 1.0
0.0 0.0 0.7 1.0
0.0 0.0 0.8 1.0
0.0 0.0 0.9 1.0
0.0 0.0 1.0 1.0
0.0 0.0 0.0 1.0
0.0 0.1 0.1 1.0
0.0 0.2 0.2 1.0
0.0 0.3 0.3 1.0
0.0 0.4 0.4 1.0
0.0 0.5 0.5 1.0
0.0 0.6 0.6 1.0
0.0 0.7 0.7 1.0
0.0 0.8 0.8 1.0
0.0 0.9 0.9 1.0
0.0 1.0 1.0 1.0
0.0 0.0 0.0 1.0
0.1 0.1 0.1 1.0
0.2 0.2 0.2 1.0
0.3 0.3 0.3 1.0
0.4 0.4 0.4 1.0
0.5 0.5 0.5 1.0
EOF
# $ECHO » running the band-structure calculation for Si…\c»
$CODE/$PW_COMMAND < si.band.$diago.in > si.band.$diago.out
# check_failure $?
# $ECHO » done»
# clean TMP_DIR
# rm -rf silicon*
# $ECHO » done»
done
Luego para generar los archivos con las bandas, hacemos el input (bands.in) para bands.x con el siguiente contenido:
—————————————–
&BANDS
prefix=’silicon’,
outdir=’.’
filband=’si.band’
lsym=.true.,
/
Para correrlo, simplemente corremos en forma serial:
bands.x < bands.in > bands.out
eso genera tres archivos : si.band.gnu, si.band y si.band.rap
El archivo gnu ya contiene las bandas en un archivo de dos columnas. Se puede obtener la gráfica en xmgr y ps usando el programa «plotband.x» de forma interactiva:
plotband.x
Input file > si.band
Reading 8 bands at 28 k-points
Range: -5.8100 16.4070eV Emin, Emax > -6.0 10.0
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.0000
high-symmetry point: 0.0000 0.0000 1.0000 x coordinate 1.0000
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 1.0000
high-symmetry point: 0.0000 1.0000 1.0000 x coordinate 2.4142
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 2.4142
high-symmetry point: 0.5000 0.5000 0.5000 x coordinate 3.2802
output file (xmgr) > si.bands.xmgr
bands in xmgr format written to file si.bands.xmgr
output file (ps) > si.bands.ps
Efermi > 6.337
deltaE, reference E (for tics) 1.0,6.337
bands in PostScript format written to file si.bands.ps

Aplicaciones con Qt
Programa «Hello world» (hello.cpp):
————————-
#include <QtGui>
int main(int argc, char **argv) {
QApplication app(argc, argv);
QLabel label(«Hello, world!»);
label.show();
return app.exec();
}
————————-
en el directorio (el nombre del directorio es el nombre del ejecutable que se genera) donde este el programa ejecutar:
$ qmake -project $ qmake $ make esto crea una aplicacione que se puede ejecutar dando click.
coordenadas de dioxido de titanio (titania)
Las dos formas usadas en fotocatálisis son la fase rutilo y la anatasa (ver http://www.chemtube3d.com/solidstate/_anatase(final).htm ).
A continuación se presentan los archivos para visualizar en xcrysden usando el comando
xcrysden –xsf archivo.xsf
!——–archivo anatasa.xsf———-
CRYSTAL
PRIMVEC
3.77600 0.00000 0.00000
0.00000 3.77600 0.00000
0.00000 0.00000 9.48600
PRIMCOORD
12 1
Ti 0.000000 0.000000 0.000000
Ti 1.888000 1.888000 4.743000
Ti 0.000000 1.888000 2.371500
Ti 1.888000 0.000000 7.114500
O 0.000000 0.000000 1.973088
O 1.888000 1.888000 6.716088
O 0.000000 1.888000 4.344588
O 1.888000 0.000000 9.087588
O 1.888000 0.000000 5.141412
O 0.000000 1.888000 0.398412
O 1.888000 1.888000 2.769912
O 0.000000 0.000000 7.512912
Por otro lado, la fase Rutilo se puede visualizar con el archivo
!——–archivo rutilo.xsf———-
CRYSTAL
PRIMVEC
4.5939982660 0.0000000000 0.0000000000
0.0000000000 4.5939982660 0.0000000000
0.0000000000 0.0000000000 2.9579973860
PRIMCOORD
6 1
Ti 0.0000000000 0.0000000000 0.0000000000
Ti 2.2969991330 2.2969991330 1.4789986930
O 1.3916051440 1.3916051440 0.0000000000
O 3.2023931230 3.2023931230 0.0000000000
O 0.9053939900 3.6886042770 1.4789986930
O 3.6886042770 0.9053939900 1.4789986930
Cálculo de espectro Raman para ZnO con Quantum Espresso
En lo siguiente se siguen las instrucciones en la dirección:
http://larrucea.eu/compute-ir-raman-spectra-qe/
En primer lugar se hace un cálculo scf creando el archivo zno.scf.in con el siguiente contenido:
!————————————–
&CONTROL
calculation = «scf»,
prefix = «ZNO»,
pseudo_dir = «./»,
outdir = «./»,
/
&SYSTEM
ibrav=0, celldm(1) =6.330582528, nat=4, ntyp= 2,
occupations=’smearing’, smearing=’gauss’, degauss=0.02,
ecutwfc =80.0, !better 140
/
&ELECTRONS
mixing_mode=’plain’
mixing_beta = 0.5,
startingwfc=’random’,
conv_thr = 1.0d-8
/
CELL_PARAMETERS alat
1.55820896 0.00000000 0.00000000
0.00000000 0.86602540 -0.50000000
0.00000000 0.00000000 1.00000000
ATOMIC_SPECIES
Zn 65.409 Zn.pbe-d-hgh.UPF
O 15.999 O.pbe-hgh.UPF
ATOMIC_POSITIONS
Zn 2.010975287 0.487933254 -0.051360548
Zn 1.234717421 0.199473387 0.448322227
O 1.051679030 0.488287222 -0.051814333
O 1.830251369 0.199830262 0.448810714
K_POINTS (automatic)
2 2 2 0 0 0
!—————————————
Para mandar correr en el cluster, enviamos el archivo qsub con
#!/bin/bash
#PBS -l nodes=1:ppn=40 -q supermicro
#PBS -N zno
#PBS -e .ERROR
#PBS -o .OUT
CODE=/usr/local/cluster_programs/espresso
cd «$PBS_O_WORKDIR»
# Run the Application
export OMP_NUM_THREADS=$PBS_NP
$CODE/pw.x < zno.scf.in > zno.scf.out
!—————————-
Luego, hacemos el cálculo de fonones con el archivo zno.ph.in con el contenido:
!————————-
Normal modes for Wurtzite
&inputph
tr2_ph=1.0d-14,
prefix=’ZNO’,
amass(1)=65.409,
amass(2)=15.999,
outdir=’.’
epsil=.false.,
trans=.true.,
asr=.true.
fildyn=’dmat.zno’
/
0.0 0.0 0.0
!—————————-
Para mandar correr hacemos el archivo ph.qsub
#!/bin/bash
#PBS -l nodes=1:ppn=40 -q supermicro
#PBS -N zno
#PBS -e .ERROR
#PBS -o .OUT
CODE=/usr/local/cluster_programs/espresso
cd «$PBS_O_WORKDIR»
# Run the Application
export OMP_NUM_THREADS=$PBS_NP
$CODE/ph.x < zno.ph.in > zno.ph.out
Finalmente, creamos el archivo de analisis zno.dm.in con el contenido:
!———————–
&input fildyn=’dmat.zno’, asr=’zero-dim’ /
!————————-
que corremos con dymat.qsub:
#!/bin/bash
#PBS -l nodes=1:ppn=40 -q supermicro
#PBS -N zno
#PBS -e .ERROR
#PBS -o .OUT
CODE=/usr/local/cluster_programs/espresso
cd «$PBS_O_WORKDIR»
# Run the Application
export OMP_NUM_THREADS=$PBS_NP
$CODE/dynmat.x < zno.dm.in > zno.dm.out
!————————————–
